
Academic Writing
Getting There First: An Evolutionary Rate Advantage for Adaptive Loss-of-Function Mutations
Biological Information: New Perspectives, edited by R. J. Marks II, M. J. Behe, W. A. Dembski, and B. L. Gordon. World Scientific Publishing, Hong Kong, 450-473.
Abstract: Over the course of evolution organisms have adapted to their environments by mutating to gain new functions or to lose pre-existing ones. Because adaptation can occur by either of these modes, it is of basic interest to assess under what, if any, evolutionary circumstances one of them may predominate. Since mutation occurs at the molecular level, one must look there to discern if an adaptation involves gain- or loss-of-function. Here I present a simple, deterministic model for the occurrence and spread of adaptive gain-of-function versus loss-of-function mutations, and compare the results to laboratory evolution experiments and studies of evolution in nature. The results demonstrate that loss-of-function mutations generally have an intrinsic evolutionary rate advantage over gain-of-function mutations, but that the advantage depends radically on population size, ratio of selection coefficients of competing adaptive mutations, and ratio of the mutation rates to the adaptive states.
Experimental evolution, loss-of-function mutations, and “the first rule of adaptive evolution”
The Quarterly Review of Biology 85(4) (December 2010), pp. 419-45.
Abstract: Adaptive evolution can cause a species to gain, lose, or modify a function; therefore, it is of basic interest to determine whether any of these modes dominates the evolutionary process under particular circumstances. Because mutation occurs at the molecular level, it is necessary to examine the molecular changes produced by the underlying mutation in order to assess whether a given adaptation is best considered as a gain, loss, or modification of function. Although that was once impossible, the advance of molecular biology in the past half century has made it feasible. In this paper, I review molecular changes underlying some adaptations, with a particular emphasis on evolutionary experiments with microbes conducted over the past four decades. I show that by far the most common adaptive changes seen in those examples are due to the loss or modification of a pre-existing molecular function, and I discuss the possible reasons for the prominence of such mutations.
Simulating evolution by gene duplication of protein features that require multiple amino acid residues
Protein Science, Volume 13, Issue 10 (October 2004), pp. 2651-2664.
Abstract: Gene duplication is thought to be a major source of evolutionary innovation because it allows one copy of a gene to mutate and explore genetic space while the other copy continues to fulfill the original function. Models of the process often implicitly assume that a single mutation to the duplicated gene can confer a new selectable property. Yet some protein features, such as disulfide bonds or ligand binding sites, require the participation of two or more amino acid residues, which could require several mutations. Here we model the evolution of such protein features by what we consider to be the conceptually simplest route—point mutation in duplicated genes. We show that for very large population sizes N, where at steady state in the absence of selection the population would be expected to contain one or more duplicated alleles coding for the feature, the time to fixation in the population hovers near the inverse of the point mutation rate, and varies sluggishly with the λth root of 1/N, where λ is the number of nucleotide positions that must be mutated to produce the feature. At smaller population sizes, the time to fixation varies linearly with 1/N and exceeds the inverse of the point mutation rate. We conclude that, in general, to be fixed in 108 generations, the production of novel protein features that require the participation of two or more amino acid residues simply by multiple point mutations in duplicated genes would entail population sizes of no less than 109.
Irreducible Complexity: Obstacle to Darwinian Evolution
In Debating Design: from Darwin to DNA, Ruse, M. and Dembski, W.A., eds. (Cambridge University Press: 2004), pp. 352-370.
Reply to My Critics: A Response to Reviews of Darwin’s Black Box: The Biochemical Challenge to Evolution
Biology and Philosophy, Volume 16, (2001) pp. 683–707.
Abstract: In Darwin’s Black Box: The Biochemical Challenge to Evolution I argued that purposeful intelligent design, rather thanDarwinian natural selection, better explains some aspects of the complexity that modern science has discovered at the molecular foundation of life. In the five years since itspublication the book has been widely discussedand has received considerable criticism. Here Irespond to what I deem to be the mostfundamental objections. In the first part of the article I address empirical criticisms based on experimental studies alleging either that biochemical systems I discussed are not irreducibly complex or that similar systems have been demonstrated to be able to evolve byDarwinian processes. In the remainder of the article I address methodological concerns, including whether a claim of intelligent design is falsifiable and whether intelligent design is a permissible scientific conclusion.
Self-Organization and Irreducibly Complex Systems: A Reply to Shanks and Joplin
Philosophy of Science 67, (2000) 155-162.
Abstract:
Some biochemical systems require multiple, well-matched parts in order to function, and the removal of any of the parts eliminates the function. I have previously labeled such systems “irreducibly complex,” and argued that they are stumbling blocks for Darwinian theory. Instead I proposed that they are best explained as the result of deliberate intelligent design. In a recent article Shanks and Joplin analyze and find wanting the use of irreducible complexity as a marker for intelligent design. Their primary counterexample is the Belousov-Zhabotinsky reaction, a self-organizing system in which competing reaction pathways result in a chemical oscillator. In place of irreducible complexity they offer the idea of “redundant complexity,” meaning that biochemical pathways overlap so that a loss of one or even several components can be accommodated without complete loss of function. Here I note that complexity is a quantitative property, so that conclusions we draw will be affected by how well-matched the components of a system are. I also show that not all biochemical systems are redundant. The origin of non-redundant systems requires a different explanation than redundant ones.
An overabundance of long oligopurine tracts occurs in the genome of simple and complex eukaryotes
Nucleic Acids Research, Volume 23, Issue 4 (February 25, 1995), pp. 689–695.
Abstract: A search of sequence information in the GenBank flies shows that tracts of 15–30 contiguous purines are greatly overrepresented in all eukaryotlc species examined, ranging from yeast to human. Such an overabundance does not occur in prokaryotlc sequences. The large Increase in the number of oligopurine tracts cannot be explained as a simple consequence of base composition, nearest-neighbor frequencies, or the occurrence of an overabundance of oligoadenosine tracts. Oligopurine sequences have previously been shown to be versatile structural elements in DNA, capable of occuring in several alternate conformations. Thus the bias toward long oligopurine tracts in eukaryotic DNA may reflect the usefulness of these structurally versatile sequences in cell function.
The protein-folding problem: the native fold determines packing, but does packing determine the native fold?
Proceedings of the National Academy of Sciences (PNAS), (May 15, 1991)
Abstract: A globular protein adopts its native three-dimensional structure spontaneously under physiological conditions. This structure is specified by a stereochemical code embedded within the amino acid sequence of that protein. Elucidation of this code is a major, unsolved challenge, known as the protein-folding problem. A critical aspect of the code is thought to involve molecular packing. Globular proteins have high packing densities, a consequence of the fact that residue side chains within the molecular interior fit together with an exquisite complementarity, like pieces of a three-dimensional jigsaw puzzle [Richards, F. M. (1977) Annu. Rev. Biophys. Bioeng. 6, 151]. Such packing interactions are widely viewed as the principal determinant of the native structure. To test this view, we analyzed proteins of known structure for the presence of preferred interactions, reasoning that if side-chain complementarity is an important source of structural specificity, then sets of residues that interact favorably should be apparent. Our analysis leads to the surprising conclusion that high packing densities–so characteristic of globular proteins–are readily attainable among clusters of the naturally occurring hydrophobic amino acid residues. It is anticipated that this realization will simplify approaches to the protein-folding problem.
Binding of p-Nitrophenyl Phosphate and Other Aromatic Compounds by β-Lactoglobulin
Journal of Dairy Science, Volume 70, Issue 2 (February 1987), pp 252-258
Abstract: Results obtained from gel filtration showed that β-lactoglobulin binds p-nitrophenyl phosphate with a stoichiometry of 1 mol of ligand per 18,360 monomer. Circular dichroic spectra confirmed the binding and implicated tryptophan and phenylalanine residues in the interaction. Fluorescence of the protein was quenched on binding also supporting complex formation; analysis of these data indicates that p-nitrophenyl phosphate binds to β-lactoglobulin A with a dissociation constant of 31 μM. The B and C genetic variants of β-lactoglobulin bind p-nitrophenyl phosphate with dissociation constants of 63 and 70 μM, respectively. In addition, a series of other nitrophenyl compounds and pyridoxal phosphate were also investigated by fluorescence analysis and found to bind to the protein. These results are discussed with respect to a recent hypothesis that β-lactoglobulin binds retinol and is structurally related to serum retinol binding protein.
Temperature‐dependent conformational transitions in poly(dG‐dC) and poly(dG‐m5dC)
Biopolymers, Volume 24, Issue2 (February 1985), pp. 289-300
Abstract: The double‐stranded helical complexes of poly(dG‐dC) and of poly(dG‐m5dC) are shown to convert from B‐ to Z‐DNA‐type conformations at moderate or low ionic strengths, lower for the 5‐methyl than for the non‐methyl species, in a highly cooperative temperature‐dependent equilibrium. In the presence of low concentrations of divalent ion, e.g., Mg2+, the temperature at which the B → Z transition occurs is virtually independent of the salt concentration and the B‐conformation is favored at lower temperature, while the Z‐conformation is favored at higher temperature. Since the Debye‐Hückel screening parameter changes rapidly with ionic strength in this region, electrostatic interaction with the free ions appears to be only a small factor in the forces that promote the transition; the temperature dependence must derive principally from effects on the solvent. The temperature dependence at high salt concentrations is also reported.
Evolution News

Darwinism Is a Potemkin Theory of Evolution

Even More Mammoth Devolution

#7 Story of 2022: Mammoth Support for Devolution
Mammoth Support for Devolution

Much Ado About Lactase Persistence
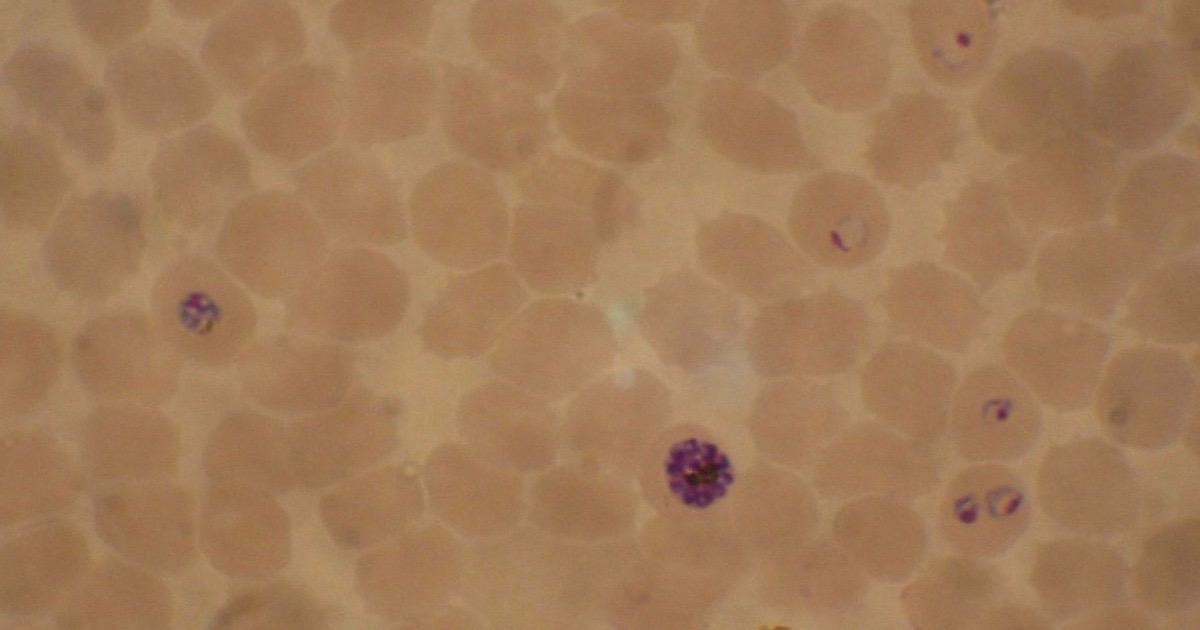
Devolution Watch: Malaria Gnaws Off a Leg
Design as a “Purposeful Arrangement of Parts”
#4 Story of 2020: Evolution, Design, and COVID-19

Excerpt: An Obstacle to Darwinian Evolution

Excerpt: A Reply to Michael Ruse

Letter to the Journal of Chemical Education

Excerpt: Darwinism and Design

COVID-19 and Biochemical Design

Citrate Death Spiral
Featured Articles

Citrate Death Spiral

A Response to My Lehigh Colleagues

Here’s How to Tell if Scientists are Exaggerating
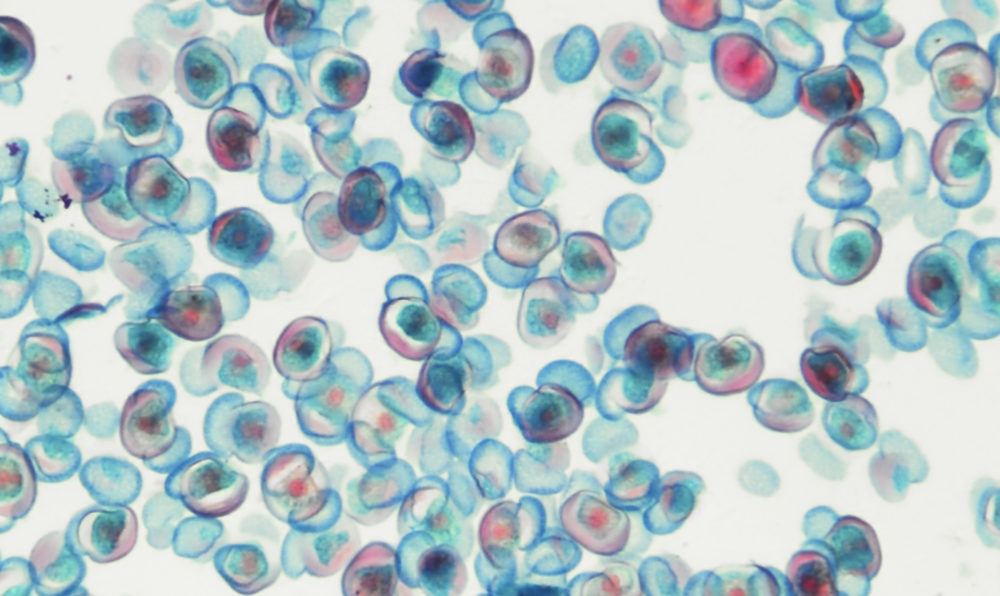
Waiting Longer for Two Mutations
A roll-up of “Waiting Longer for Two Mutations”, Parts 1-5
Science, E. coli, and the Edge of Evolution
Whether Intelligent Design is Science
A Response to the Opinion of the Court in Kitzmiller vs Dover Area School District
Michael Behe on Molecular Exploitation and the Theory of Irreducible Complexity

Design for Living
The Basis for a Design Theory of Origins
In Defense of the Irreducibility of the Blood Clotting Cascade
Response to Russell Doolittle, Ken Miller and Keith Robison
“A True Acid Test”
Response to Ken Miller
A Mousetrap Defended
Response to Critics
Teach Evolution
And Ask Hard Questions
Molecular Machines
Experimental Support for the Design Inference
The Sterility of Darwinism

Darwin Under the Microscope
